Add and modify an ELN entry
Introduction
This view allows creating and editing an electronic laboratory notebook entry (reactions).
User preferences
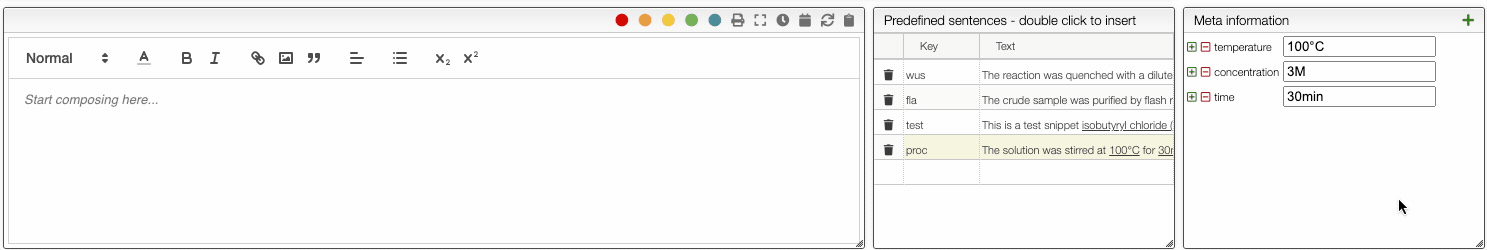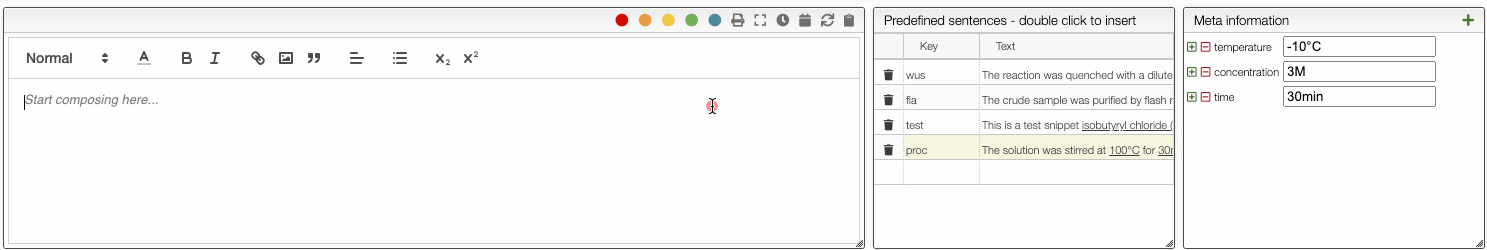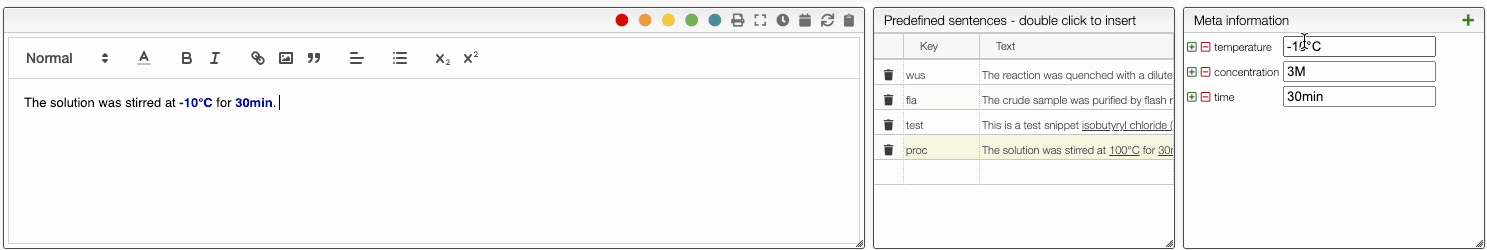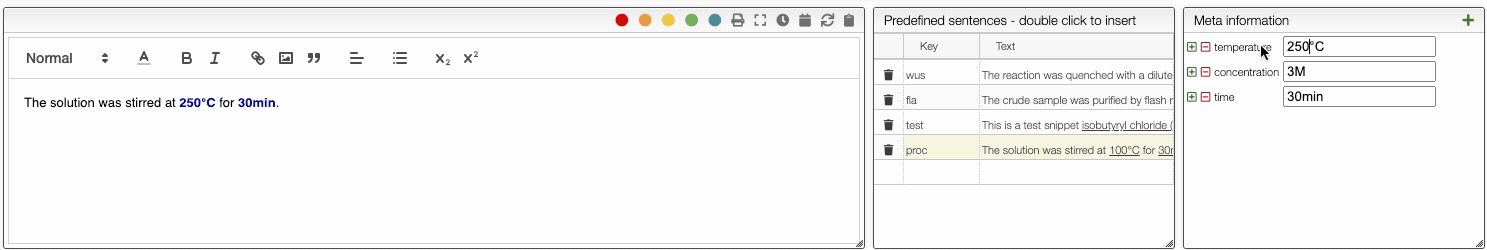
To define user preferences and snippets you should click on the Prefs button.
Predefined sentences
![]()
To define a snippet first associate to it a key that is composed of lowercase letters and then define the corresponding sentence. The sentence may contain not only reagents like r1, r2 but also meta information that can be inserted using _ followed by the meta field name like _temperature. If a snippet contains those fields you will be able to update their values automatically.
Each snippet should be associated with a key composed of lowercase letters.
You can then insert snippets by typing their key that will be automatically replaced and updated with the current values.
PubChem lookup and autoupdate
You can also define if you would like to have automatic safety lookup in PubChem as well as automatic updates of the reagents and meta information that has been inserted in the procedure.
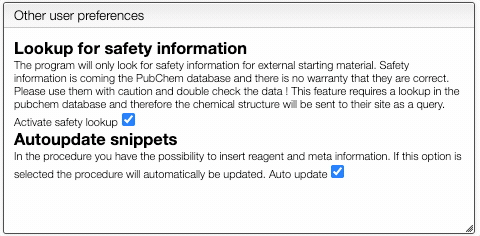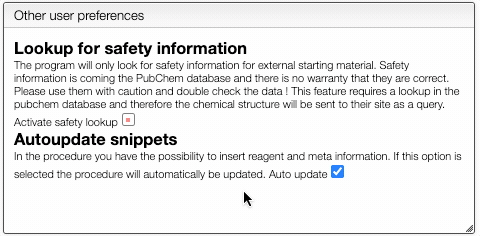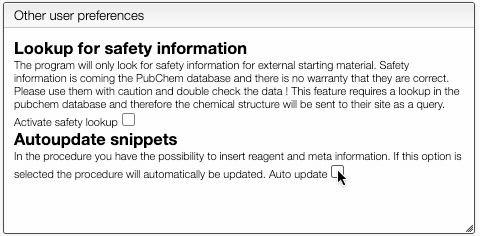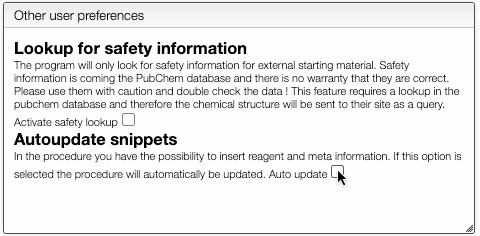
Don't forget to Save preferences!
Organic reaction
When preparing a new organic reaction the idea is first to add the different reagents and then to fill the schema with simple clicks. The concept behind it is that either the product is commercially or it was synthesized before. Therefore, all the chemical structures of the reagents are already known.
Lookup for a product
You should use the code column to look up a product.
In this column you may either enter:
- CAS number
- product name
- molecular formula
- product code (practical to retrieve a product you synthesized before)
A molecular formula may be entered the way a chemist think about it. Meaning you are allowed to use groups and parenthesis like Me2CHCOCl.
Once the string is entered, press tab to trigger the lookup. The system will search a reference database of 400,000 molecules as well as all the internal products you have access to.

Click on the right product to copy the name, structure and density.
Defining the quantities
The reagent calculator is connected to databases and can retrieve information about a chemical. For instance in the "code" you may enter a molecular formula, name or CAS number and the system will look for commercially available chemicals. You can then select the molecule you want to add in the table.
- If you enter a new sample and change the molecular formula, molecular weight will be automatically calculated. In the molecular formula you may enter groups like Et, Ph, Ts, ... as well as parentheses.
- The purity may be entered in
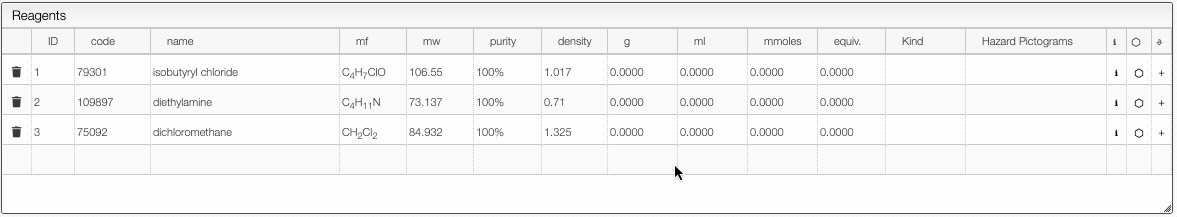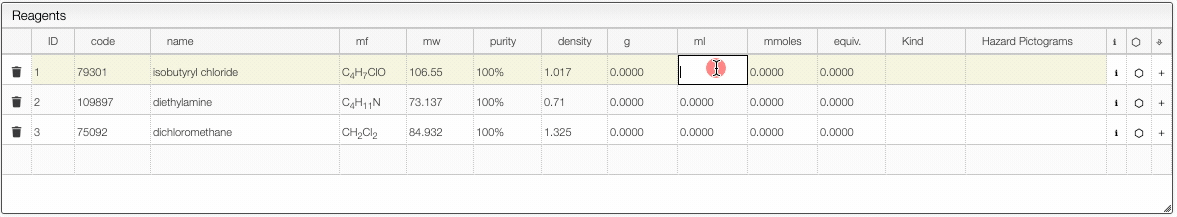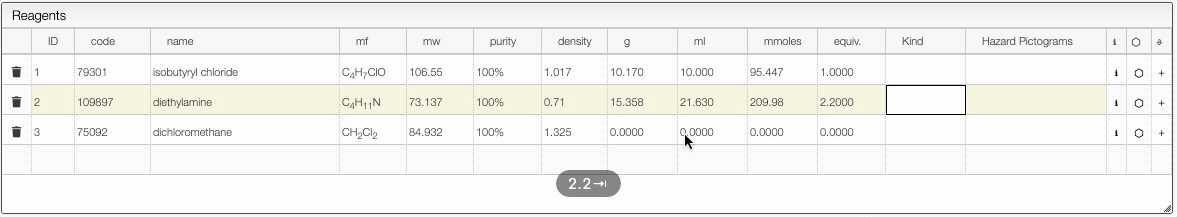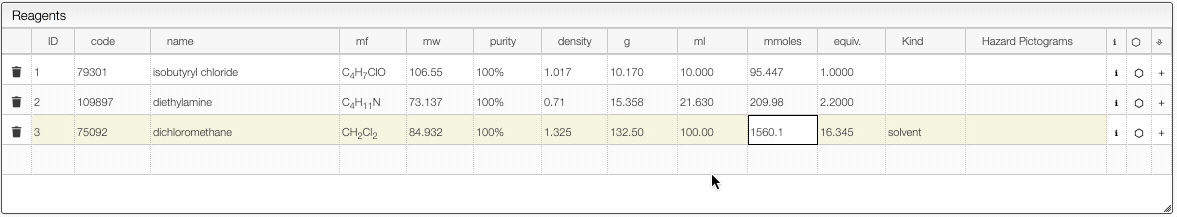
%,M(molar),mM(millimolar) orL(loading). Molar is expressed in mmoles / mL and loading is expressed by mmoles / g and is practical for solid phase synthesis. - The first reagent for which you add a quantity will be defined as 1 equivalent. You may still change this anytime. After defining the first quantity you will probably want to define the other reagents from the equivalent columns rather than the quantity (except probably for the solvent).
- Once the equivalents are specified, the samples are “connected”. This means that if you change the quantity of one reagent, all quantities will change.
- It is possible to remove the link between the reagents by unselecting the “Link” checkbox.
Drawing the schema
JSME is a simple-to-use and powerful tool developed by Peter Ertl and Bruno Bienfait. In order to draw the reaction we will start by adding the chemical structures from the reagent table.
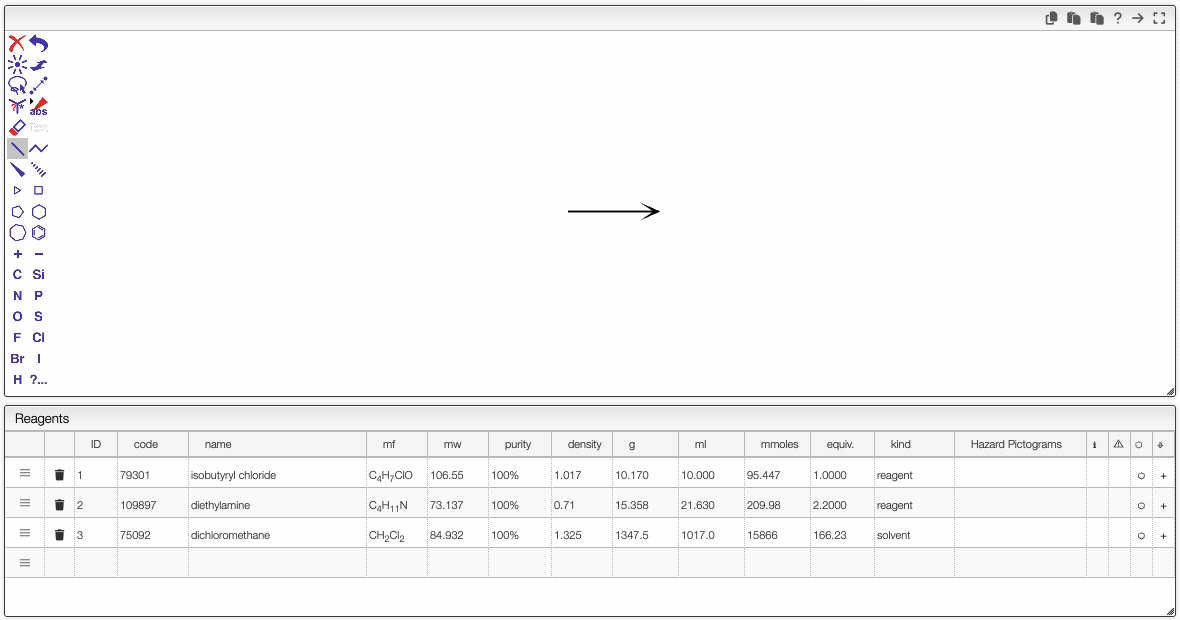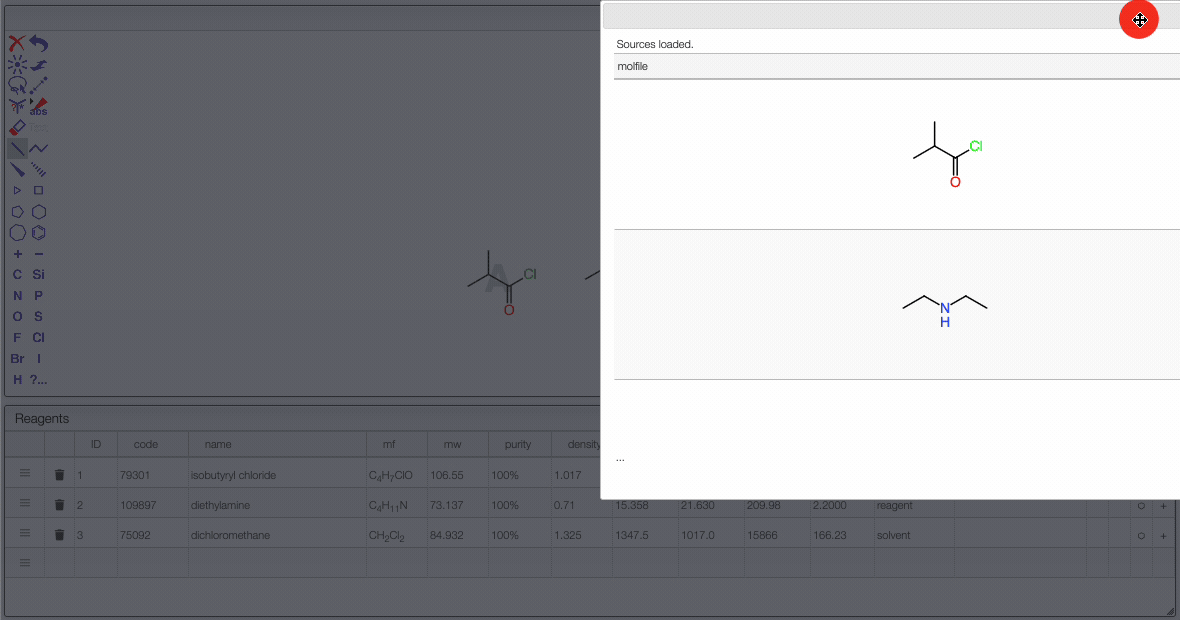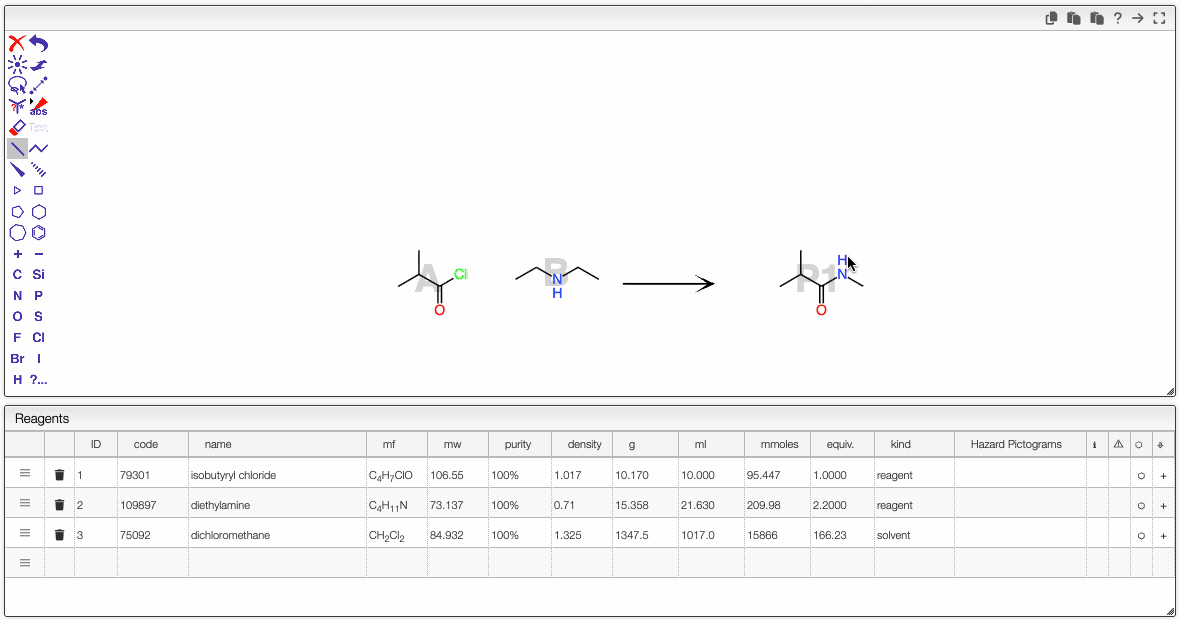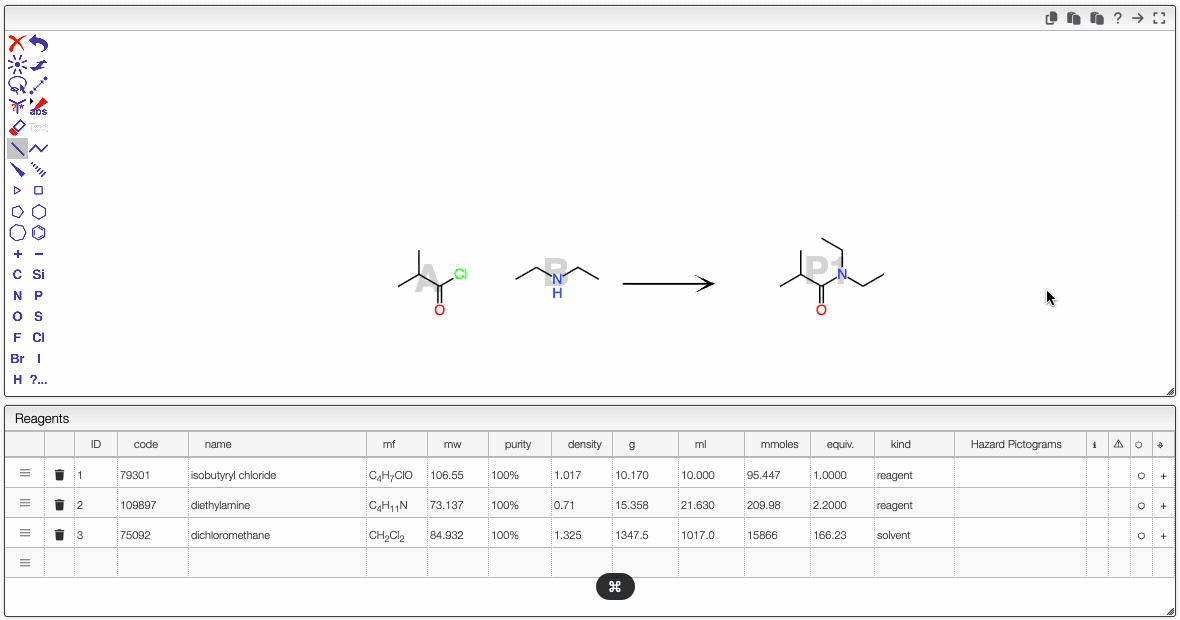
It is possible to select a molecule by hovering over it. Once selected, you can move it (just click around and drag the mouse).
Even if the molecule does not appear to be selected, you can still act on it. For instance, you can:
- copy the molecule to the other side of the reaction by clicking on the arrow.
- click on the white rectangle to delete this selected molecule.
Check analysis
A reaction contains various samples. Samples can be either isolated and purified products or any analysis related to the reaction.
A sample contains all the analyses related to it (NMR, GC, IR, Mass, etc.) and each analysis has a specific view to process it.
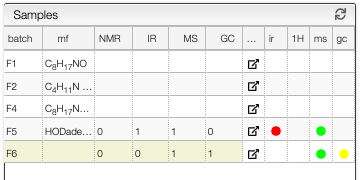
It is now possible to jump directly to the corresponding analysis view by clicking on the corresponding color bullet from the sample list.
To create a new sample, you simply need to click on the button Add empty sample and choose a batch name, this will create a new batch where you can include any type of spectra and additional information. Once you refresh the page in the global reaction, you will see the new batch in the list. If you click on the batch, there will be a preview of the information you have entered.
Some tips for organic reactions
You can specify the purity of a reagent.
Specifying the purity of a reagent
The purity of a reagent may be specified using 3 possible units:
%: purity as mass ratio- like 40% HNMe2 in water
M(ormM): moles/liter (or mmoles/liter)- like 1.6M BuLi
L: loading: mole/kg- useful for solid phase synthesis
?: unknown purity- when working with natural products like wood or in materials science, the sample does not have a molecular formula, and therefore it is not possible to define the number of mmoles. Using
?as units allows you to freely define weight, density, and volume.
- when working with natural products like wood or in materials science, the sample does not have a molecular formula, and therefore it is not possible to define the number of mmoles. Using
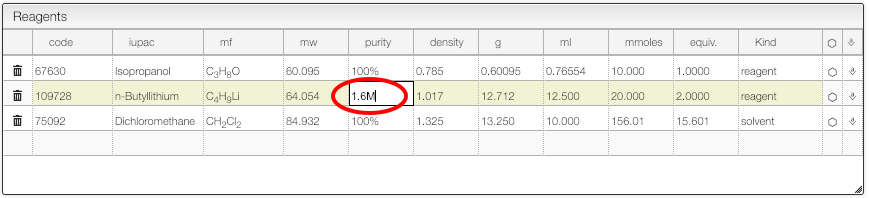
The system will calculate continuously the molecular formula of all the reagents and products.
Products theoretical information
When you draw products of reaction the application will calculate continuously the expected quantities for 100% and one equivalent.
This value is also updated when you change the quantities in the reagent table.

There is an easy way to deal with mixtures of solvents.
Calculate mixture of solvents volume to reach specific concentration
For some reactions it is important to calculate the solvent to reach a specific concentration and in some cases it can be a mixture of solvents.
The reagent table allows dealing with mixture of solvent. In this case we would like to work in DMSO/H2O in a ratio (70/30):
- For the solvents enter the number of equivalent so that the sum is
1 - Enter the concentration you want to achieve in the 'Purity' column of all the solvents.
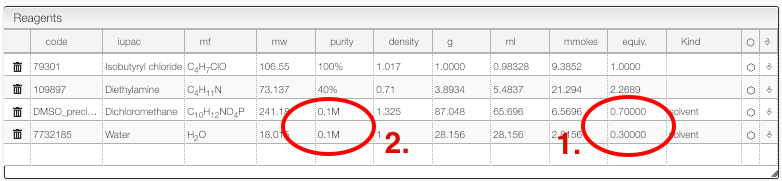
In some cases it is required to add many times the same reagent.
Same reagent multiple times
In some reactions such as in the example below, the product is formed via the combination of multiple reagent molecules.
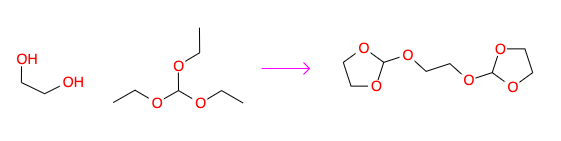
In this example the mixture of ethylene glycol with triethyl orthoformate yields a cyclic orthoester. In order to enter the quantities for this kind of reaction you need to specify correctly the number of equivalents of the reagent that limits the yield. In this case the triethyl orthoformate limits the yield, so we enter two equivalents for this reagent.
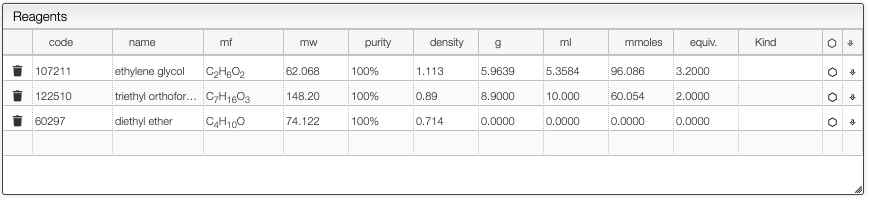
This will allow calculating correctly the expected quantity of the product as well
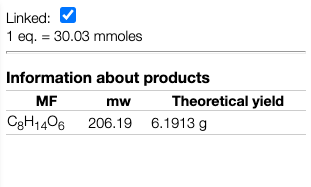
In some cases it is required to copy manually the structure of a reaction product.
Copy product as molfile
If a sample was created using a method other than from the reaction, the chemical structure, molecular formula, and molecular weight will not be copied automatically. This is the case, for example, when a sample is analyzed by NMR and the sample is created automatically during import.
There is a simple way to copy / paste the chemical structure of the product.
The product is copied as a molfile and can be paste in a sample entry.
It could be pasted for example in ChemDraw using Edit -> Paste Special -> Mol text
Insert snippets, reagents and meta information
It is possible to define an unlimited number of snippets and to recall them directly in the procedure by typing its abbreviation (followed by Tab, Space, or Enter).
All the reagents can also be inserted by entering r followed by the reagent number. i.e. r2.
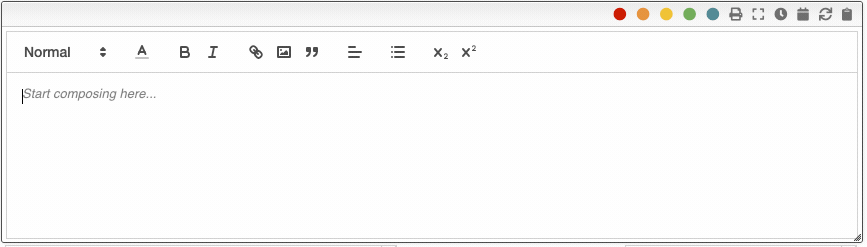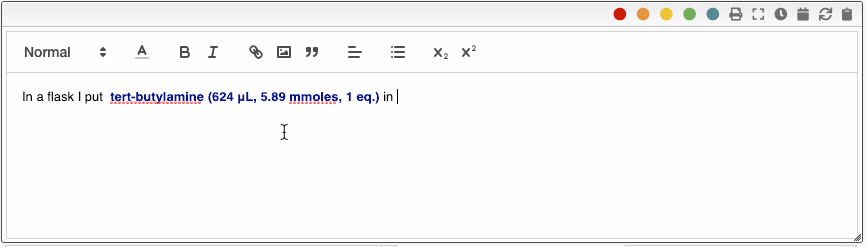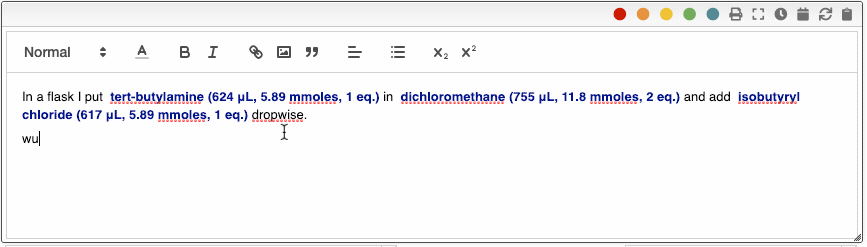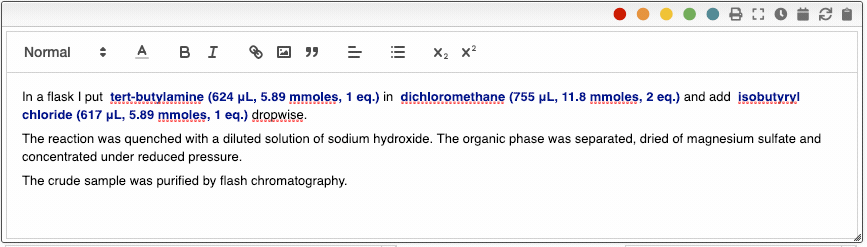
The ELN also allows you to add an unlimited number of meta information key-value pairs. This meta information can be inserted not only in the procedure but also in snippets. To include meta information, type _ followed by the name of the property, for example _temperature. Using meta information allows you to centralize all the parameters that change from one reaction to another. This is especially useful in the case of parallel synthesis.
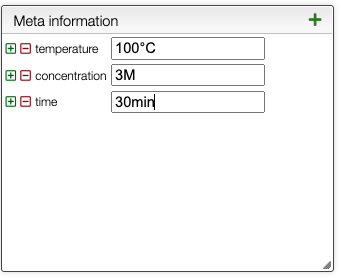
It is also possible to define default meta information, that can be reused in any new or old reaction. To retrieve the default meta information, just click on the ![]() button.
You can add at any time labels to the list of default meta information in the following panel.
button.
You can add at any time labels to the list of default meta information in the following panel.

Insertion of predefined sentences
In order to insert predefined sentences you can click on the clipboard icon.
A dialog containing the list of predefined sentences will appear, and you should
click on the one you want.
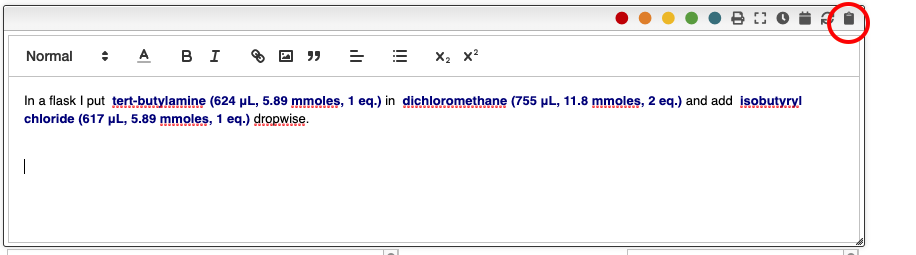
Miscellaneous
Drag drop images
You may just copy / paste an image of the “Drop or paste” zone, and it will be inserted in the procedure.
Calculate solvent volume to reach specific concentration
For some reaction it is important to calculate the solvent to reach a specific concentration.
The reagent table allows to do this:
- For the solvent enter as number of equivalent '1'
- Enter the concentration you want to achieve in the 'Purity' column.
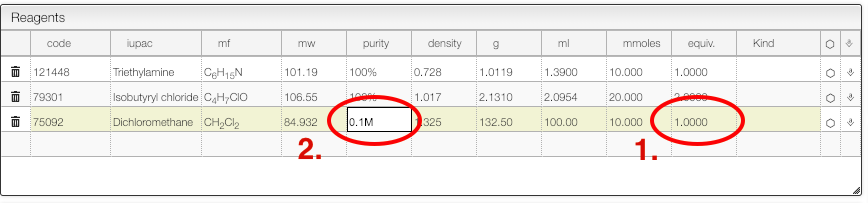
Safety information
When looking for commercial products you may decide to also lookup for safety information in PubChem.
To activate this feature, click on prefs and check Activate safety lookup. Don't forget to save your new preferences!
It is also possible to directly edit the safety information by clicking in the Hazard pictogram column and entering the various GHS pictogram code separated by a comma.

More information from PubChem about the chemical can also be found by clicking on the 'i' icon.
The safety information can be updated by clicking on the ![]() icon. This will automatically update the safety information from PubChem.
icon. This will automatically update the safety information from PubChem.