Search by NMR chemical shifts
This tool is designed to search by NMR chemical shifts, which can be useful for finding similar pure products or identifying products in complex mixtures such as in metabolomics. To select the chemical shifts, you may either enter the values directly in the Ranges to search table or ALT + click directly on the spectrum.
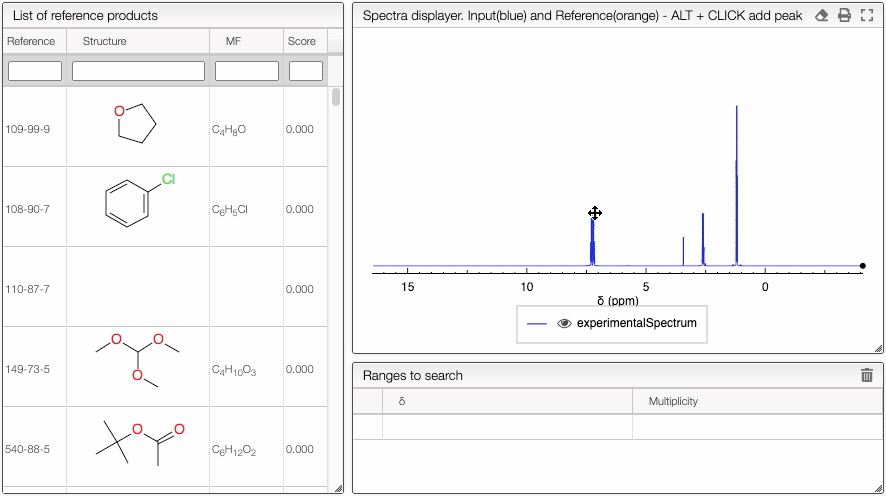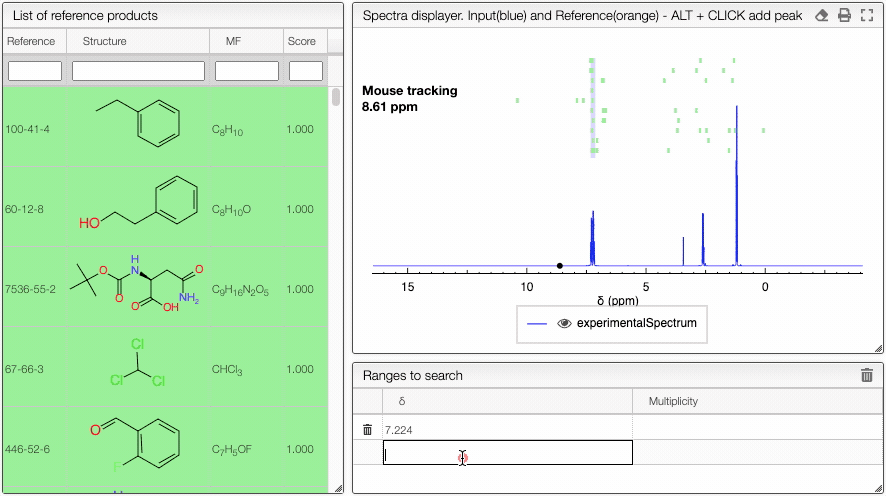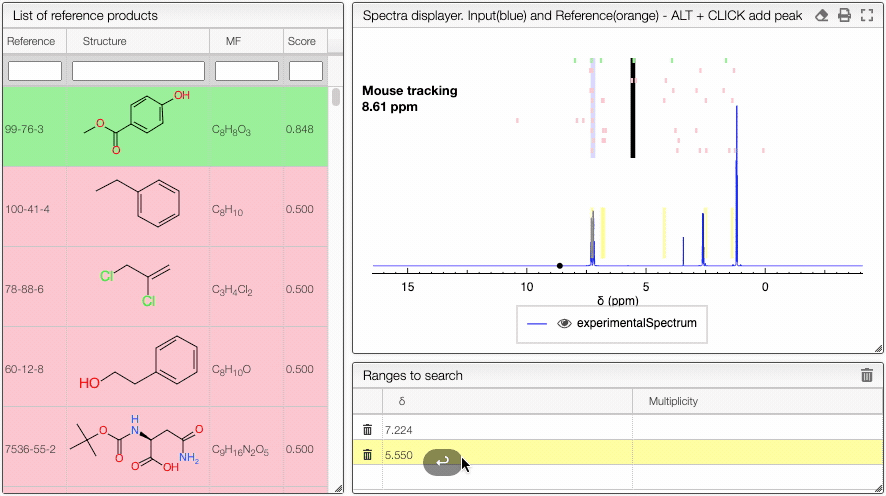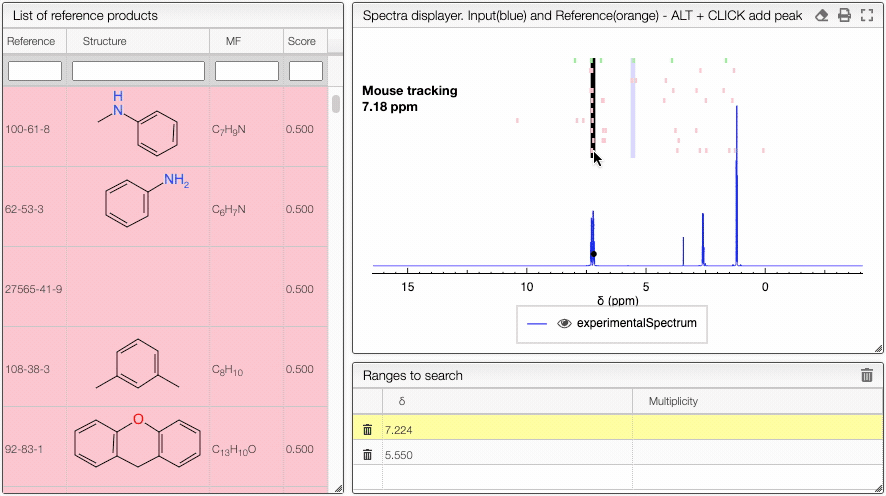
You may at any time either delete one of the lines by clicking on the ![]() icon in front of the row, or you may delete all the chemical shifts by clicking on the same icon on the top right of the table. You may as well specify the multiplicity. The score will receive some bonus if the multiplicity matches (and can therefore have a score over one).
icon in front of the row, or you may delete all the chemical shifts by clicking on the same icon on the top right of the table. You may as well specify the multiplicity. The score will receive some bonus if the multiplicity matches (and can therefore have a score over one).
Adding an experimental spectrum
Experimental spectrum can be added either by clicking on Load demo, by dragging & dropping a JCAMP-DX file or by selecting an existing spectrum from the database. Those spectra will be filtered based on the Exclusion zones.
How is a JCAMP-DX file structured.
JCAMP-DX file format
JCAMP-DX (Joint Committee on Atomic and Molecular Physical Data Exchange) is a standard file format for the exchange of spectra and related physical and chemical information between different spectrometers, databases or other systems.
The information is stored using ASCII characters and the file can be viewed, corrected, and annotated with a text editor. The spectra are stored as a table containing (x,y) coordinate pairs. Besides the data points, it is possible to store meta information and comments. The file extension is .jdx.
A JCAMP-DX document is composed of an unlimited number of Labelled Data Records (LDRs). Each LDR starts with a “##” and ends with “=”. Any space, comma, slash or hyphen is removed and the text is written with capital letters.
Some examples of Data Labels:
- TITLE : title of the experiment
- END : the last line of the file
- XUNITS : the units reported on the x-axis
- NPOINTS : number of points
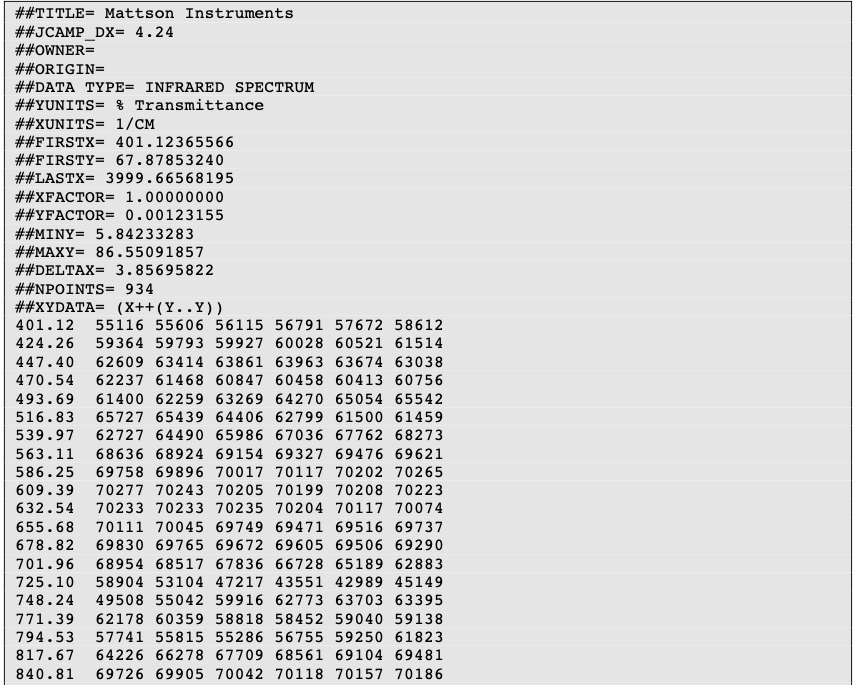
Two important LDRs are “XYDATA” and “PEAKTABLE”, which contain the spectral information. The former gives information in the form of a table where the first value in a line stands for an x coordinate and any subsequent values are y-coordinates with an equidistant increment on the x-axis. The latter provides information as a collection of (X,Y) pairs.
It is commonplace to compress the data tables. For instance, the table of numbers can be replaced by a line of characters (pseudo-digits). Among these pseudo-digits, there are PAC, SQZ, DIF, DIFDUP.

An example of compressed data using DIFDUP

An in depth description is given in the original paper by McDonald and Wilks. Insofar as JCAMP-DX is a well-described and accessible format, it partially aligns with the FAIR (Findable, Accessible, Interoperable, Reusable) principles . It is interoperable and reusable. Provided that the user makes it findable and accessible, JCAMP-DX will fully comply with the aforementioned principles.
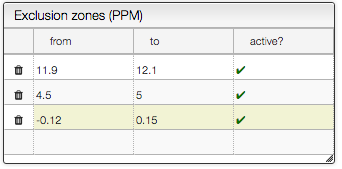
The exclusion zones will be indicated on the spectrum by a red line.

Specify the allowed error and display the result
While searching for similar spectra the program will allow an error on the chemical SHIFT ⇧. This error is specified in:

Each peak will be represented as an allowed zone by a blue rectangle.

And the signals of the 10 best matches will be represented as 10 different lines. The color will represent the score:
- light green: > 0.8
- orange : 0.6 < score <= 0.8
- pink : score 0.4 < score <= 0.6
Hovering over the annotations will highlight the corresponding molecule. If you click on the annotation, you will get the detailed information.
Interactive results
Hovering over the list of reference products will highlight the region where peaks are expected.
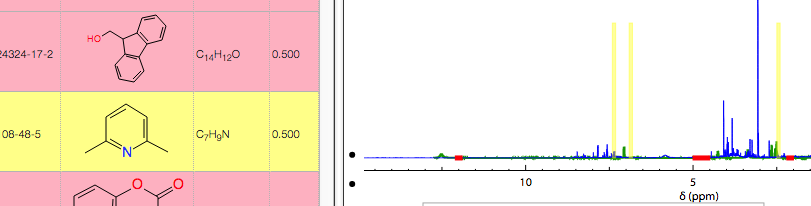
This allows you to quickly browse through the list of reference spectra. If one of the references looks promising, just click on it to display the corresponding reference NMR spectrum.
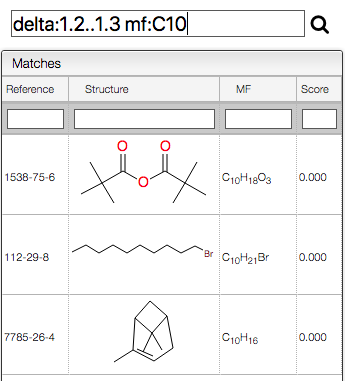
Quick search
You may enter complex queries to search for reference products. This includes searching by molecular formula, chemical SHIFT ⇧, and reference.
You may for example search for products that have a chemical SHIFT ⇧ between 10 and 11:
delta:10..11
Or combine queries for the chemical shifts as well as the molecular formula containing 10 carbons:
delta:2..2.1 delta:3..3.4 mf:C10
The spectra displayer
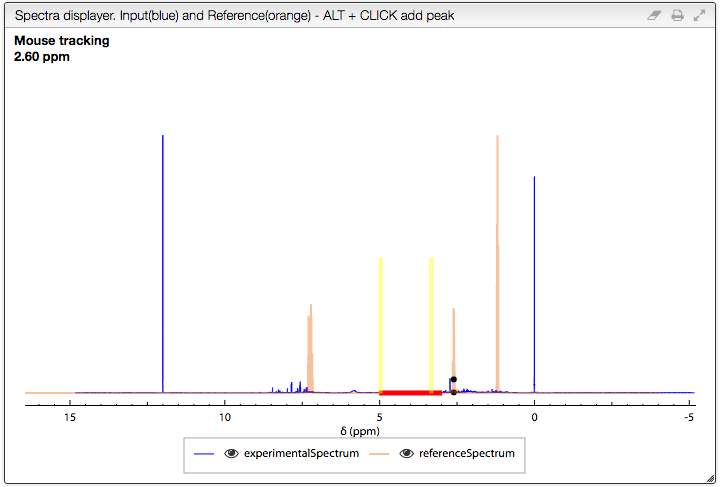
The following options are available in the spectra displayer:
- click + drag + release : zoom in the spectrum
- double click: zoom out = display the full spectra
SHIFT ⇧+ double click : zoom out progressively- scroll wheel : vertical scale
- select a series (click on the corresponding line in the legend) + scroll wheel : vertical scale of a specific series
- hide a series by clicking on the corresponding eye in the legend