Mass fragmentation
Simulation of mass fragmentation
Analysis of mass fragments of small molecules obtained for example by electronic impact may be cumbersome.
To facilitate the process, this tool allows you to draw a molecule and select the breakable bonds. The system will then generate all the possible fragments and recombine them.
Select the ionization method
In order to observe a mass spectrum the molecule has to be charged and this ionization has an impact on the mass. You may enter a list of ionizations that was applied to the molecule in the following box:

For instance for electronic impact you would enter a simple ‘+’. Meaning that we have removed an electron to charge the molecule.
Draw / edit the molecule
You may either draw a molecule directly in the editor or paste a molfile coming from another software. For instance if you have a molecule in ChemDraw™ you may select the molecule and ‘Edit -> Copy as … -> MOL Text’. Then you may put the mouse over the drawing applet and press ‘CTRL + V’ (on windows) or ⌘ + V’ on mac.

If you would like to draw efficiently the molecules directly in the editor click on the little question mark and try to learn all the shortcuts.
JSME
JSME Help and Basic Instructions
JSME Menu
![]() shows molecule SMILES
shows molecule SMILES
![]() clears the editing area (or the current molecule, if structure contains several parts)
clears the editing area (or the current molecule, if structure contains several parts)
![]() starts new molecule component (when multipart option or reaction input are enabled)
starts new molecule component (when multipart option or reaction input are enabled)
![]() delete mode (click on atom or bond to delete it)
delete mode (click on atom or bond to delete it)
![]() deletes functional group - choose this option and then click bond connecting the group with the main skeleton
deletes functional group - choose this option and then click bond connecting the group with the main skeleton
![]() starts atom numbering or reaction mapping (when this option is enabled)
starts atom numbering or reaction mapping (when this option is enabled)
![]() query atom / SMARTS (when this option is enabled)
query atom / SMARTS (when this option is enabled)
![]() switches between allowed charged states on clicked atom
switches between allowed charged states on clicked atom
![]() reaction input (when reactions are enabled)
reaction input (when reactions are enabled)
![]() undo
undo
![]() redo
redo
![]() next ring will be added as a spiro ring
next ring will be added as a spiro ring
![]() version information and link to the help page
version information and link to the help page
![]() input stereo bond, click existing stereo bond to cycle through possible types (up, down, orientation)
input stereo bond, click existing stereo bond to cycle through possible types (up, down, orientation)
![]() powerful chain tool, allows creation of various chains and rings; click single bond to change it to double
powerful chain tool, allows creation of various chains and rings; click single bond to change it to double
![]() allows input of inorganic atoms or atoms in nonstandard valence state (in this case enter the atom symbol enclosed in [], for example the carbene carbon will be [C])
allows input of inorganic atoms or atoms in nonstandard valence state (in this case enter the atom symbol enclosed in [], for example the carbene carbon will be [C])
![]() opens functional group popup menu
opens functional group popup menu
![]() move selected atom useful when drawing crowded structures (visible when enabled in options)
move selected atom useful when drawing crowded structures (visible when enabled in options)
Basic Editing
Rings are added connected by a single bond (when not added to a terminal atom); if you want to add a spiro ring press SHIFT ⇧ when adding it. An alternative way to add a spiro ring (that may be used also on touch devices) is to press the ![]() icon; the next ring will be then added as spiro.
icon; the next ring will be then added as spiro.
With single bond selected, a click on existing single bond changes it to double.
With the powerful chain tool ![]() you can draw chains, saturated rings and by clicking on single bond change it to a double bond.
you can draw chains, saturated rings and by clicking on single bond change it to a double bond.
Molecule may be moved by "dragging" free space and rotated by pressing also the SHIFT ⇧ key.
JSME Keyboard Shortcuts
If you are using JSME often, learn the following shortcuts. They will speed-up your editing considerably.
ESC returns to the standard editing state (carbon, single bond)
d or Del starts delete mode
atoms C, N, O, P, S, F, L (for Cl), B (for Br), I, H, R
bonds - for single bond, = for double bond
rings 3..8 for 3 to 8 membered rings, 1 for phenyl and 0 for furyl
groups a - COOH, y - NO2, z - SO3H, t - tert. butyl, ft - CF3
Stereochemistry
Stereochemistry at C4 centers, double bonds and allenes is supported. Use the up / down wedge bonds to indicate stereochemistry at the C4 centers. Remember, that only bonds with a "sharp point" towards the atom are considered. When creating SMILES the editor tries to guess missing stereo features, in unresolvable cases an error message is issued and the SMILES without stereo information is created.
When the "autoez" option is set, SMILES with E,Z stereochemistry on all non-ring double bonds will be generated automatically. Without this keyword (or for ring double bonds) you have to mark a double bond as stereo by clicking on it with the stereo bond button selected. The bond color will change to magenta.
Stereochemistry may be completely disabled by the "nostereo" option.
Atom Numbering
Atom numbering (marking) is enabled by the option "number" (for reaction input this is default). Button ![]() appears in the menu. To mark an atom the "numbering mode" must be on, selected by clicking the 123 button. The atom number is chosen by pressing respective keys on your keyboard (i.e. 2 or 12); then atom(s) must be clicked. To delete the number pres 0 at your keyboard and then click the atom.
appears in the menu. To mark an atom the "numbering mode" must be on, selected by clicking the 123 button. The atom number is chosen by pressing respective keys on your keyboard (i.e. 2 or 12); then atom(s) must be clicked. To delete the number pres 0 at your keyboard and then click the atom.
Input of Multipart Structures
By default only non-disconnected structures may be entered. This may be changed by a "multipart" option. A button ![]() appears in the menu. A new molecule may be started only after clicking this button, then selecting a proper template (atom, ring or bond) and clicking free space in the drawing area. Without clicking the NEW button first clicking the free space has the same effect as in the standard mode (i.e moving or rotation of the last touched molecule). In the multipart mode the
appears in the menu. A new molecule may be started only after clicking this button, then selecting a proper template (atom, ring or bond) and clicking free space in the drawing area. Without clicking the NEW button first clicking the free space has the same effect as in the standard mode (i.e moving or rotation of the last touched molecule). In the multipart mode the ![]() button deletes the last touched molecule.
button deletes the last touched molecule.
Reaction Input
Reaction input is enabled with the option "reaction". Buttons ![]() ,
, ![]() and
and ![]() appear in the menu and arrow appears also in the drawing area. Now simply draw reactant(s), product(s) and modulator(s) (modulators have to be above the arrow) as explained in the description of input of multipart structures. The arrow button enables simplified input of reactions. After clicking it, the reactant will be copied to the product (including atom numbering, if any).
appear in the menu and arrow appears also in the drawing area. Now simply draw reactant(s), product(s) and modulator(s) (modulators have to be above the arrow) as explained in the description of input of multipart structures. The arrow button enables simplified input of reactions. After clicking it, the reactant will be copied to the product (including atom numbering, if any).
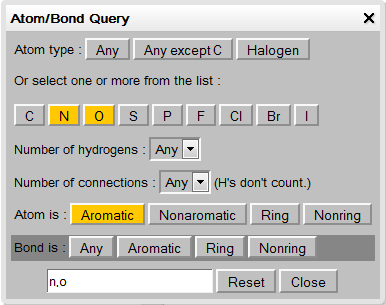
Query Features
Query button ![]() (when enabled in the options) launches a query window that allows creation of SMARTS atom or bond queries by combining various atom attributes. Select the SMARTS options in this panel and then click the respective atom or bond in the molecule.
(when enabled in the options) launches a query window that allows creation of SMARTS atom or bond queries by combining various atom attributes. Select the SMARTS options in this panel and then click the respective atom or bond in the molecule.
Copy and Paste
Right mouse click opens the Copy and Paste pop-up menu that allows to copy the molecule as SMILES or MOL file or paste MOL , SDF, or RXN files into the editor.

On supported touch devices, the menu can be opened by �touch and hold� on the surface of the editor.
Keyboard shortcuts Ctrl C (for copy) and Ctrl V (for paste) are supported on most browsers.
Drag and Drop of Files in the Editor
Some HTML 5 compatible browsers support the drag and drop of files. Starting with version 2013-03-24, JSME accepts dropping of text files in the format MOL, SDF and RXN.
![]()
To drag molecule from the editor into the appropriate destination (for example Word or text document, or desktop) drag the icon in the lower right corner of the editor.
Define the cleavable bonds
In the drawing applet you should select the blue bullet icon and click on the bonds that are breakable.
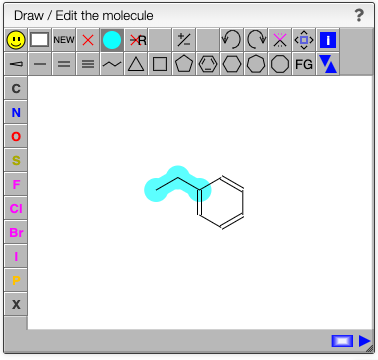
The fragments with their corresponding mass will be calculated on the fly as well all the possible recombinations of those fragments. This may be useful to determine unknown side products of a reaction.
Analyze the results
A virtual spectrum with all possibilities is generated, and hovering over an annotation on a peak will highlight the parts of the molecule required to reach that mass.
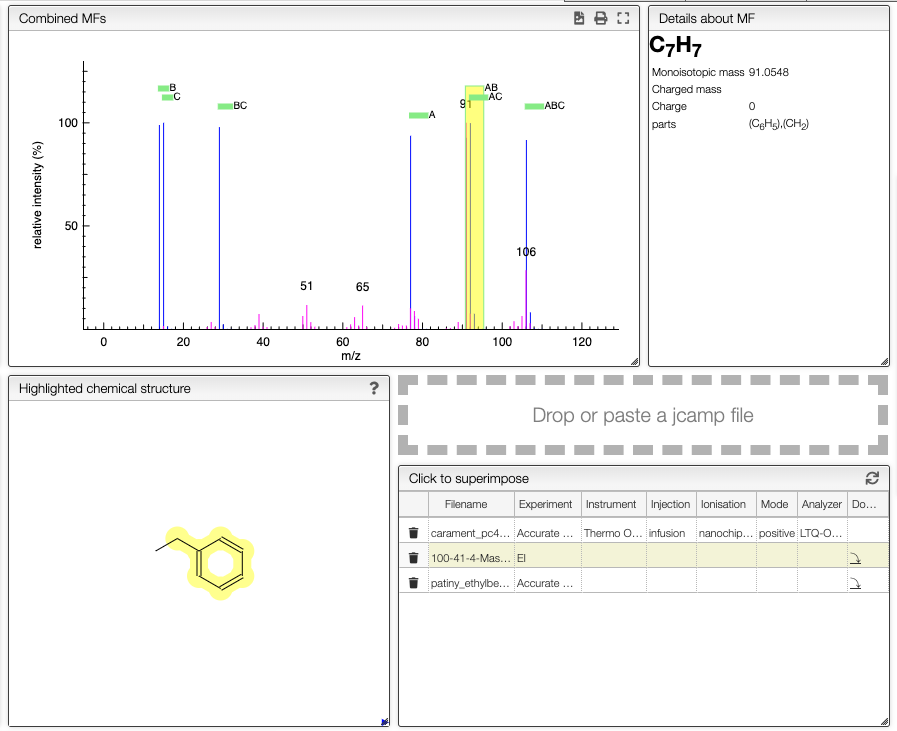
If you have a XY text file or JCAMP-DX containing an experimental spectrum you may directly drag and drop or paste on the drop zone.
You may also have mass spectra saved in the database and one click on the name will superimpose the spectrum to the predicted one.
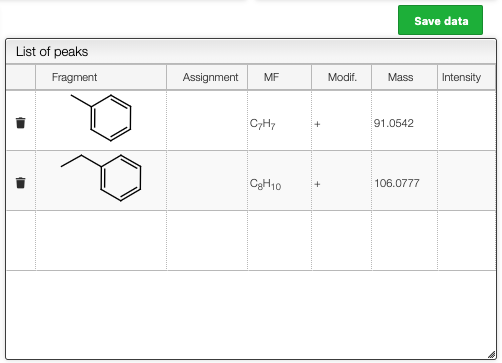
Assign fragments
The list of possible recomposed molecules also contains the intensity if there is an experimental spectrum.
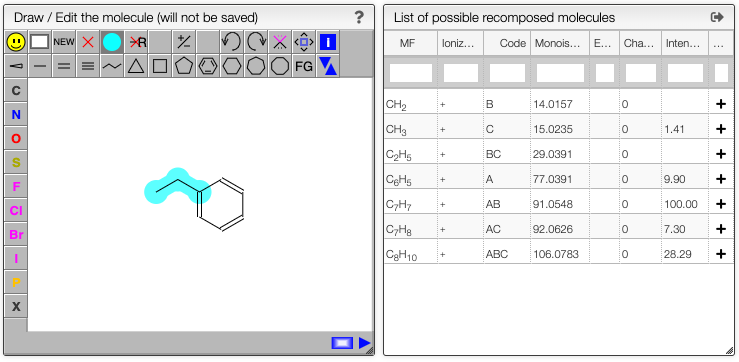
For this list clicking to the '+' on a line will add this fragment in the list of peaks.
The same result can be achieved by pressing ALT + click on an annotation in the spectrum.
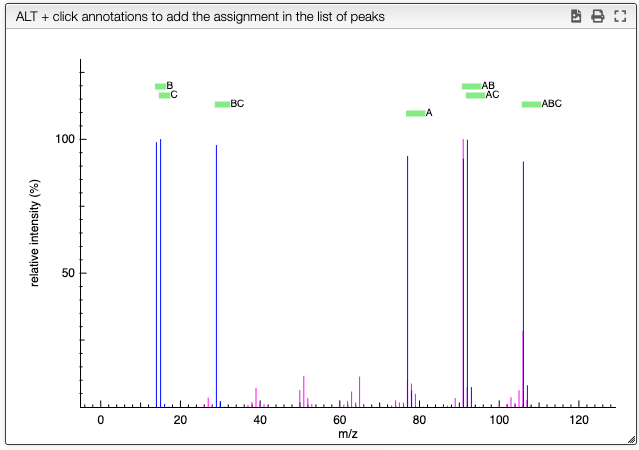
The list of assigned peaks can then be saved in the database by clicking on Save data.