Analyze high resolution mass spectra of complex mixtures
Introduction
This tool allows you to analyze complex mass spectra of complex mixtures including polynucleotides or proteins/peptides that may involve digestion, adducts, residue modifications, etc.
Importing experimental data
This tool can be either used in a LIMS or stand-alone. In the stand-alone mode you should either drag/drop your experimental spectrum as a tab-delimited text file or copy and paste it (CTRL-V) while moving you mouse over the drop zone.

The list of available spectra will be displayed in the table, and you may click on one of them to display it.
Parameters
Ionizations
All the results are shown for the neutral molecule, and it is therefore necessary to specify the ionization method. The methods may be entered as a comma separated list of all the allowed ionizations.
You may as well specify multiple charges. Here are some examples of allowed patterns:
- H+ (addition of a proton)
- H+, Na+, K+ (either a proton, sodium cation or potassium cation)
- H+, (H+)2, (H+)3 (addition of one, two or three protons)
- (H+)-1, (H+)-2, (H+)-3 (removal of one, two or three protons)
It is also allowed to enter a range
- (H+)1-10 (addition of one to 10 protons)
- (H+)-1--10 (removal of one to 10 protons)
Mixture
This tool allows you to generate possible molecular formulas based on the MF range.
The tool is a combination of all the lines and for each line one molecular formula will be used. If there are many possibilities you can separate on each line the molecular formula using a comma. Moreover you can define ranges of molecular formula.
Example:
H (CH2CH2O)0-2 OH, H (CH2CH2S)0-2 SH
H-1Me, H-1Ph,
The first line will define 6 possible molecular formula. The second line means remove an hydrogen followed by adding a Me or Ph. Please note the ending comma, which means that there may also be no modification.
We can then also allow many ionizations like for example H+, Na+ and the following table will be obtained:
| First part | Second part | Ionization | MF Mass | m/z |
|---|---|---|---|---|
| HOH | H+ | H2O | 19.0178 | |
| HOH | H-1 Me | H+ | CH4O | 33.0335 |
| HOH | Na+ | H2O | 40.9998 | |
| HOH | H-1 Me | Na+ | CH4O | 55.0154 |
| H(CH2CH2O)OH | H+ | C2H6O2 | 63.0441 | |
| H(CH2CH2O)OH | H-1 Me | H+ | C3H8O2 | 77.0597 |
| H(CH2CH2S)OH | H+ | C2H6OS | 79.0212 | |
| H(CH2CH2O)OH | Na+ | C2H6O2 | 85.026 | |
| H(CH2CH2S)OH | H-1 Me | H+ | C3H8OS | 93.0369 |
| HOH | H-1 Ph | H+ | C6H6O | 95.0491 |
| H(CH2CH2O)OH | H-1 Me | Na+ | C3H8O2 | 99.0417 |
| H(CH2CH2S)OH | Na+ | C2H6OS | 101.0032 | |
| H(CH2CH2O)2OH | H+ | C4H10O3 | 107.0703 | |
| H(CH2CH2S)OH | H-1 Me | Na+ | C3H8OS | 115.0188 |
| HOH | H-1 Ph | Na+ | C6H6O | 117.0311 |
| H(CH2CH2O)2OH | H-1 Me | H+ | C5H12O3 | 121.0859 |
| H(CH2CH2O)2OH | Na+ | C4H10O3 | 129.0522 | |
| H(CH2CH2S)2OH | H+ | C4H10OS2 | 139.0246 | |
| H(CH2CH2O)OH | H-1 Ph | H+ | C8H10O2 | 139.0754 |
| H(CH2CH2O)2OH | H-1 Me | Na+ | C5H12O3 | 143.0679 |
| H(CH2CH2S)2OH | H-1 Me | H+ | C5H12OS2 | 153.0402 |
| H(CH2CH2S)OH | H-1 Ph | H+ | C8H10OS | 155.0525 |
| H(CH2CH2S)2OH | Na+ | C4H10OS2 | 161.0065 | |
| H(CH2CH2O)OH | H-1 Ph | Na+ | C8H10O2 | 161.0573 |
| H(CH2CH2S)2OH | H-1 Me | Na+ | C5H12OS2 | 175.0222 |
| H(CH2CH2S)OH | H-1 Ph | Na+ | C8H10OS | 177.0345 |
| H(CH2CH2O)2OH | H-1 Ph | H+ | C10H14O3 | 183.1016 |
| H(CH2CH2O)2OH | H-1 Ph | Na+ | C10H14O3 | 205.0835 |
| H(CH2CH2S)2OH | H-1 Ph | H+ | C10H14OS2 | 215.0559 |
| H(CH2CH2S)2OH | H-1 Ph | Na+ | C10H14OS2 | 237.0378 |
Nucleic and peptidic sequence
There are various possibilities to enter a peptidic sequence
One letter code
AAAHHHCCCKK
A sequence may be entered as a one letter code. By default the system will add on the N-terminal side an hydrogen
H and on the C-terminal side a OH.
N or C terminal modifications should be entered between parenthesis. For example if you have an amide function on
the C-terminal you should enter AAAHHHCCCKK(NH2).
It is also possible to have side chain modifications by adding the modification after the one letter amino acid between parenthesis.
Examples:
AAAC(S-1Se)GGG: the sulfur of a cysteine is replaced by a selenium (S-1, we remove a sulfur,Sewe add a selenium)AAA(H-1Cl)GGG: we replace the hydrogen of an alanine by a chlorineAAAS(PO3H): we have a phosphorylated serine
Three letter code
Sequence may be entered as a 3 letter code. In this case lowercase / uppercase has to be respected and N and C terminal groups must be specified.
Examples:
HAlaGlyProOHMeAla(Se)GlyNH2(CH3)Ala(Se)GlyNH2
Peptides
In the case of peptide it is also possible to in-silico digest the sequence and to specify the allowed missed cleavage and the minimal and maximal sequence length.
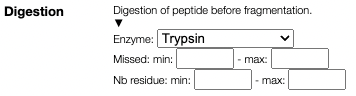
While the usual fragmentation types ABCXYZ are allowed, it is also possible to specify internal fragments like YA and YB.

This may lead to numerous sequences that will have to be processed, and if you would like to focus on a part of the sequence you can use the "Sequence must contain" field. This allows you to enter a string that must be contained in the expanded sequence code.
This means that if you have a sequence AGSTY and would like to only process the fragments that contain the glycine you should enter in this field Gly or if you want only ST you should enter SerThr.
Sequence must contain field also allows as value a regular expression. In this case you can enter something like /ProVal|gly/i.
You need to enter the / to specify it should be considered as a regular expression and you have the possibility to specify modifiers like in this case in which we want a case insensitive match (i).
Nucleotides
Links
It is possible to create links inside the sequence of between sequence and variable groups. Links are defined using #n where n is a number. When generating all the possible molecular formula there should exactly 2 times each #n (or zero time) otherwise the MF is not valid.
You will find here after 2 examples.
Cyclic peptides
You can enter as sequence:
AC(H-1#1)LLIIC(H-1#1)KK
For the 2 cysteines we remove a proton and create a link between them.
Variable modifications
If we have a cyclic peptide that is linked by a polyethylene glycol of variable number of units we can enter:
- sequence: AC(H-1#1)LLIIC(H-1#2)KK
- variable groups: #1(CH2CH2O)1-10#2,
For the molecular formula to be valid, #1 and #2 must either be absent (zero occurrences) or each present exactly twice. This is especially useful in peptide fragmentation.
Report
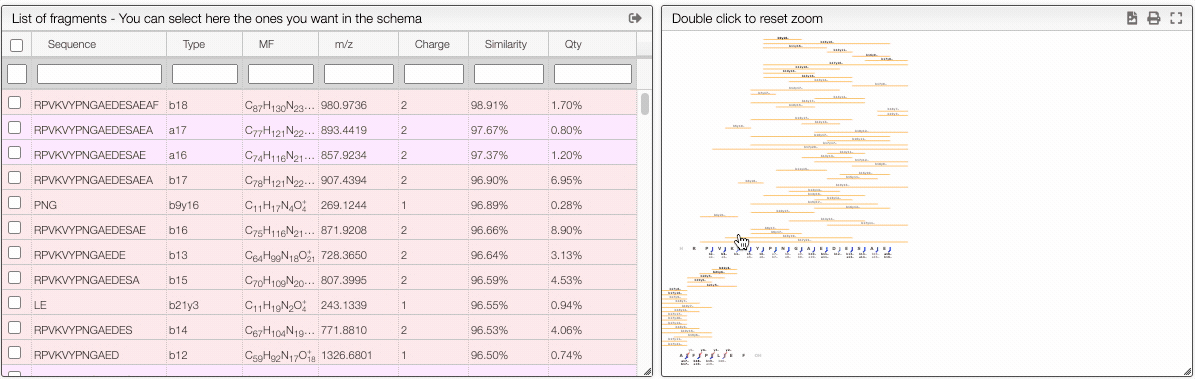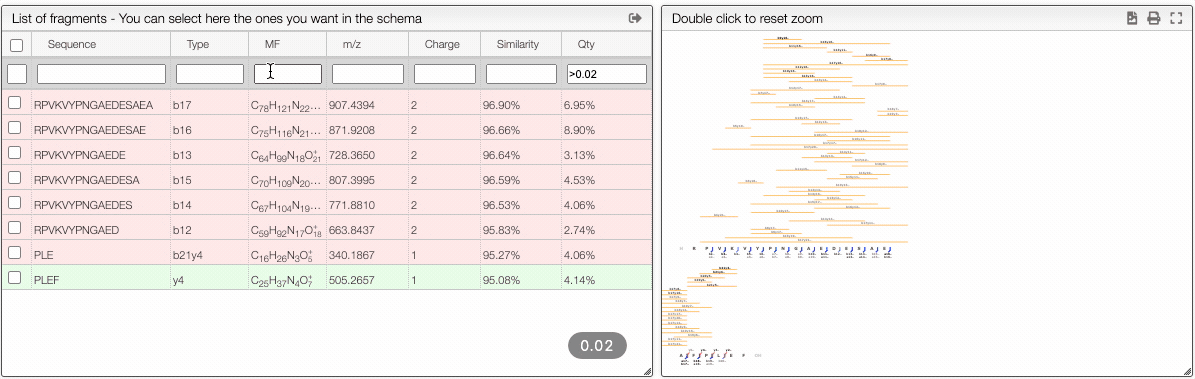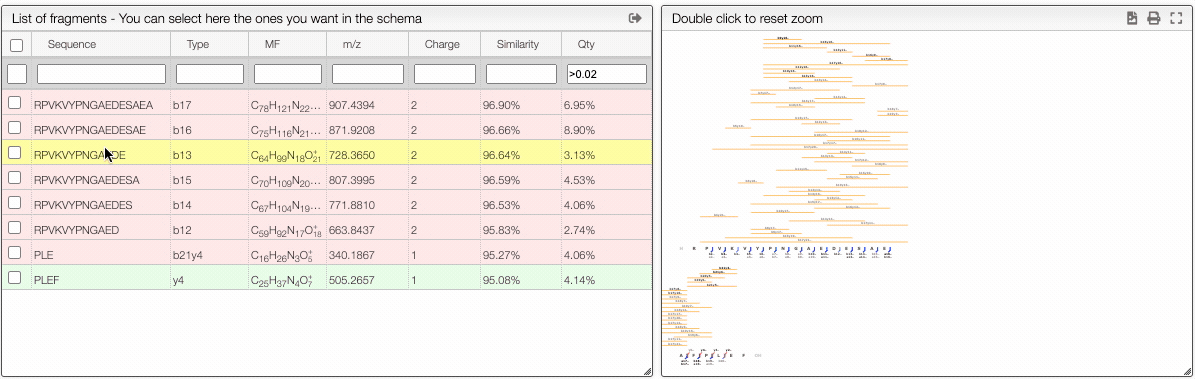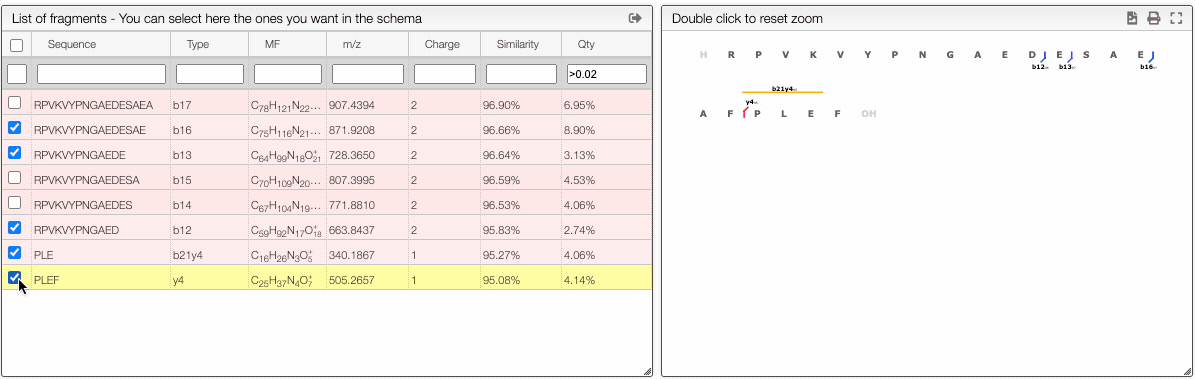
The report tab allows you to create a dynamic image that shows the various fragmentation patterns observed for peptide and nucleic acid sequences.
It is a vectorial format (SVG) suitable for publication and preferences allows to define various rendering parameters.
There are also 2 ways to filter the displayed fragments.
Dynamically filter fragment
The more visual way is based on interactive preferences that allow you to show or hide internal fragments as well as to select the minimum similarity and minimum quantity of fragments to display.
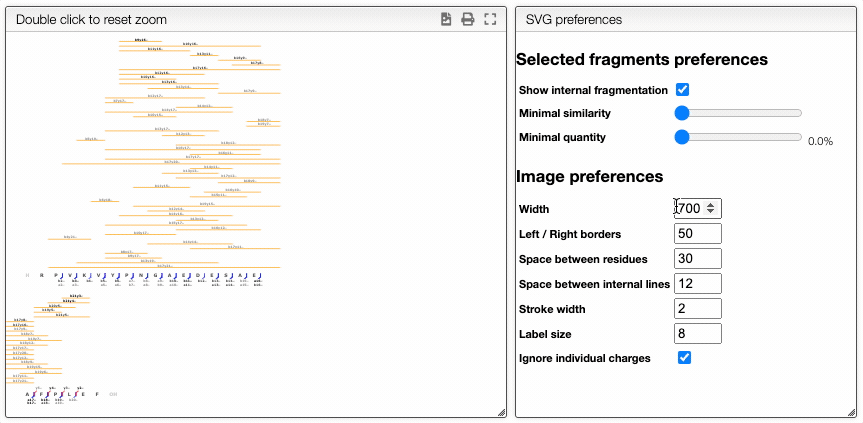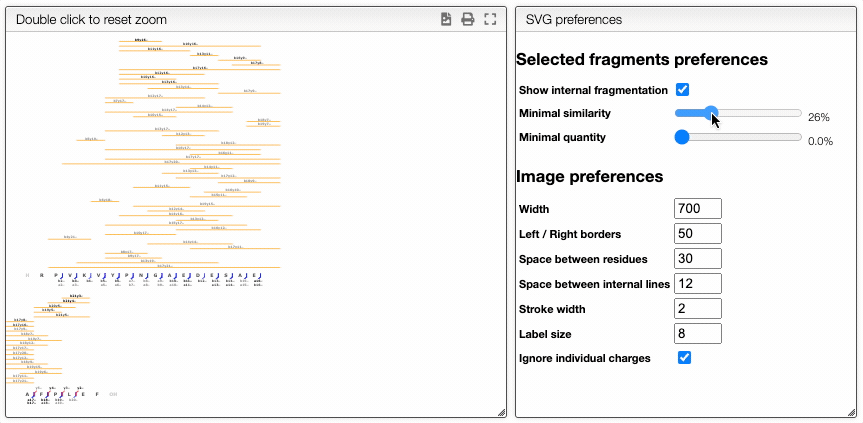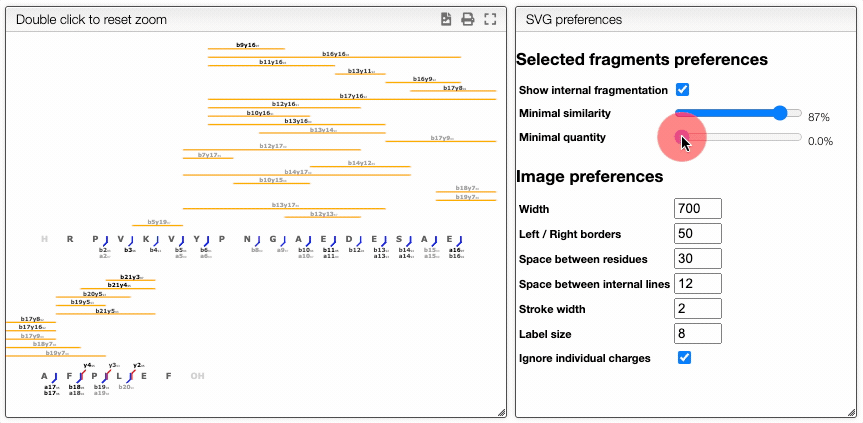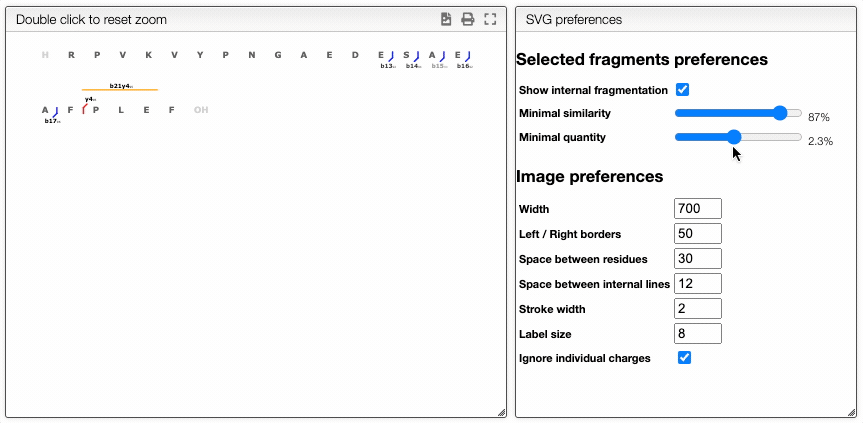
Filtering from the table
From the table it is also possible to make queries per column. To display only the selected fragments, click on the top of the select column. It is also possible to select a few fragments to display.